14 Tools for High-throughput data
14.1 High-throughput genomic data
Sequence Read Archive
SRA SRA (Sequence Read Archive)는 High-throughput 시퀀싱 데이터의 공개 데이터베이스 중 가장 큰 규모의 미국 국립 보건원(NIH)의 1차 데이터베이스로서 서열데이터 뿐만 아니라 메타데이터, 유전체, 및 환경 데이터를 포함합니다. NCBI와 EBI(European Bioinformatics Institute), DDBJ(DNA Database of Japan) 간 국제적 제휴를 통해 세 기관에서 제출 받은 데이터는 서로 공유되고 있습니다.
간략한 사용법은 NBK569238 또는 SRA download 문서 이곳을 참고하시기 바랍니다.
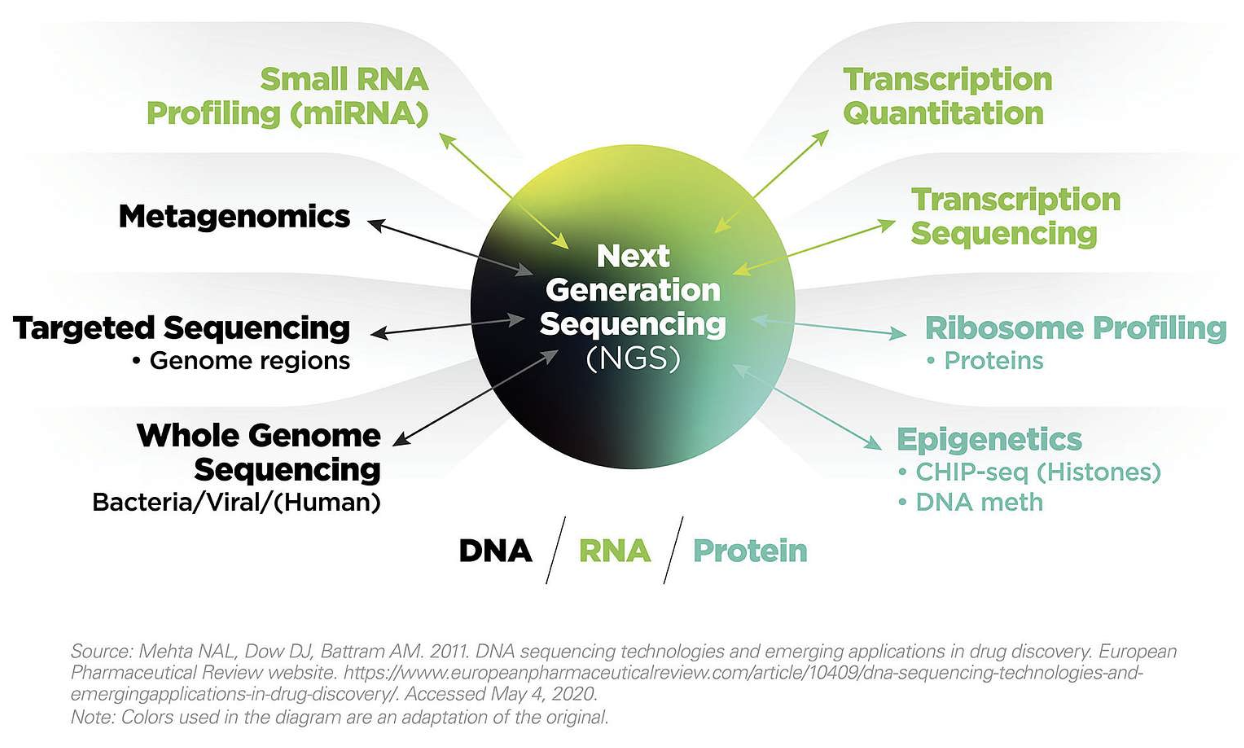
데이터를 다운로드 할 수 있는 NCBI SRA Toolkit을 제공하며 이 중 MS Windows 64 bit architecture 를 다운로드 받아 압축을 풀고 사용할 적절한 디렉토리로 옮겨 줍니다. 여기서는 D:\sratoolkit.3.0.0-win64이 곳에 이동해 두었고 전체 디렉토리 구성은 다음과 같습니다.
명령을 어느 디렉토리에서나 사용하고 싶다면 위 경로의 bin 디렉토리를 path로 잡아주는 과정이 필요합니다. 다음 위치로 이동 후 “내PC > 속성 > 고급 시스템 설정 > 환경변수” 를 클릭하면 다음 창이 생성됩니다.
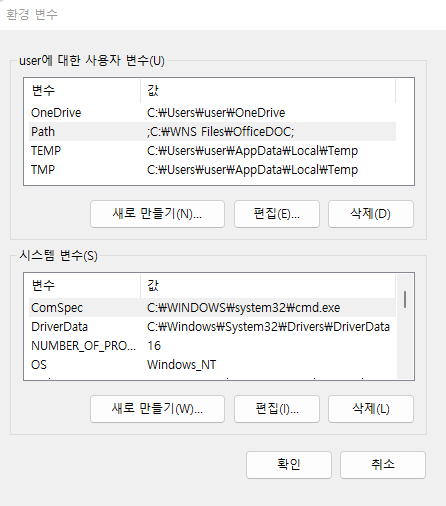
Path를 선택후 편집을 클릭하면 다음 화면이 생성되고 새로만들기를 누른 후 D:\sratoolkit.3.0.0-win64\bin라고 입력해주고 모든 창에서 확인을 눌러주면 되겠습니다.
이제 파일 탐색기로 파일을 다운로드 받을 작업 디렉토리로 이동한 후 주소창에 cmd이라고 입력해서 프롬프트가 있는 명령창을 실행합니다.
fastq-dump.exe를 사용해서 다운로드 받을 수 있으며 최근에는 fasterq-dump를 사용해서 더욱 빠르게 다운로드를 받을 수 있습니다.
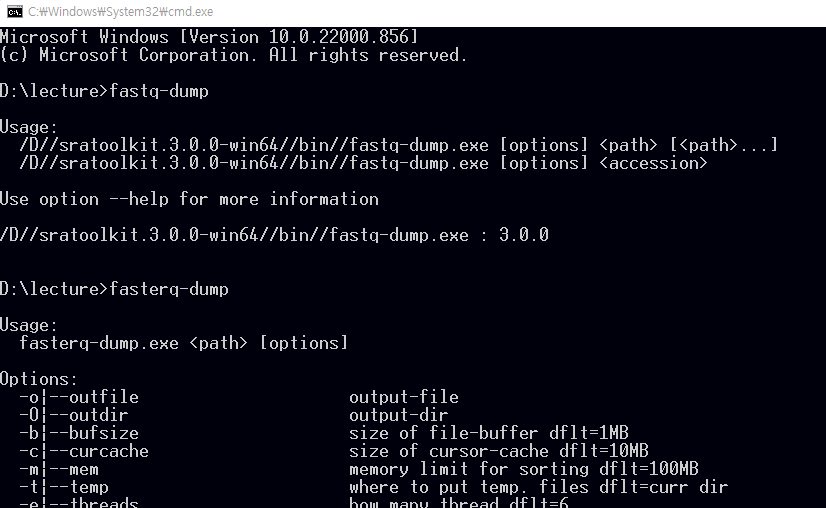
뒤에서 설명할 GEO 데이터베이스에서 GSE148719 데이터를 다운로드 해보겠습니다. 위 링크를 클릭해서 들어가면 화면 하단의 SRA Run Selector 라는 링크가 있고 이를 클릭하면 다음과 같은 화면이 보입니다.
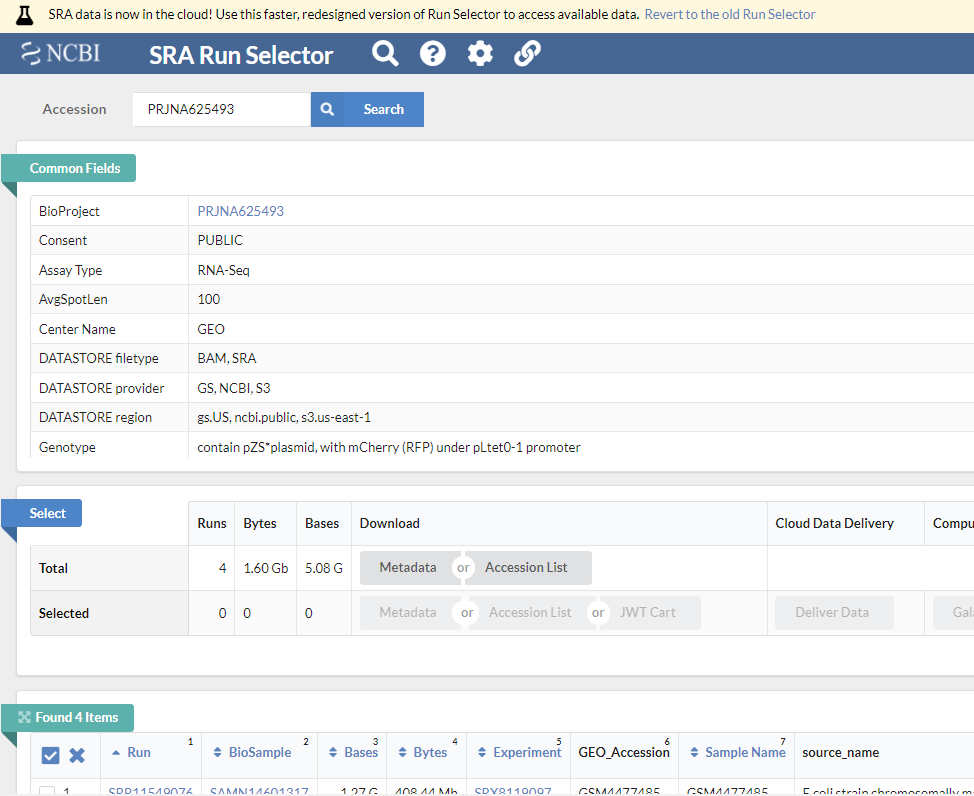
Metadata (SraRunTable.txt) 와 Accession list (SRR_Acc_List.txt)를 파일 형태로 다운로드 받은 후 적절한 전처리 후 사용하면 되겠습니다.
prefetch --option-file SRR_Acc_List.txt만약 하나의 fastq 데이터만 다운로드 받을 경우 다음과 같습니다.
prefetch SRR11549076이후 fasta 파일로 변환해 줍니다
fasterq-dump --split-files SRR11549076100000개 read만 별도로 저장
fastq-dump -X 100000 --split-files SRR11549076Gene expression omnibus (GEO)
GEO는 microarray, next-generation sequencing 등의 high-throughput 유전체 데이터를 보유한 공공 저장소입니다.
- 대규모 기능유전체 데이터베이스
- 데이터 기탁 쉽게 만들고 고수준 QC 유지
- 사용하기 쉬운 인터페이스 유지
Platform, Sample, Series로 구성되어 있으며 Platform은 사용된 어레이 플랫폼에 대한 설명과 데이터 테이블로 구성되어 있습니다. GPLXXX 형태의 GEO 액세스 번호가 할당되며 하나의 플랫폼은 많은 샘플들에 사용될 수 있습니다. Sample은 개별 샘플이 처리된 조건 등의 설명이 있는 테이블로 구성되며 GSMxxx 형태의 GEO 등록 번호가 할당됩니다. Sample은 하나의 Platform만 참조 가능하며 여러 Series에 포함될 수 있습니다. Series는 관련된 샘플을 그룹화하고 전체 연구의 주요 설명을 제공합니다. GEO 등록 번호 GSExxx가 할당됩니다.
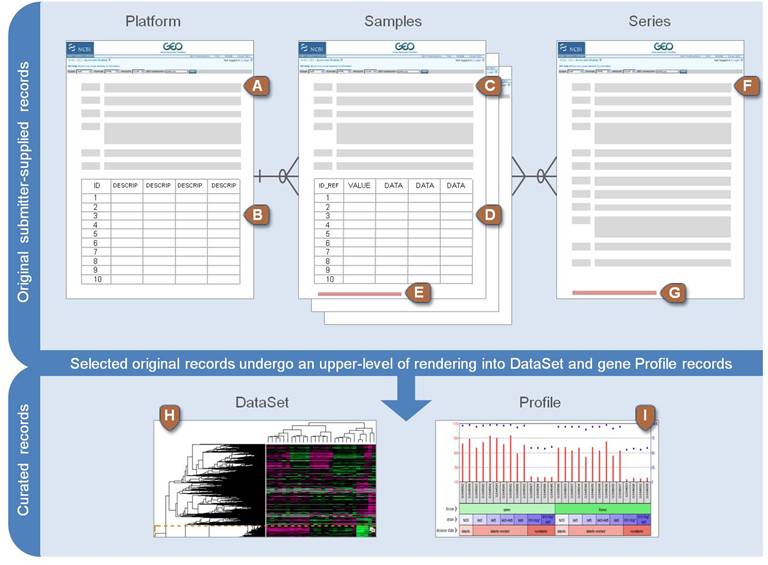
위 세 가지 타입 외에 Datasets 이 있으며 Datasets은 GDSxxx 아이디를 가집니다. 앞서 Series (GSExxx) 데이터가 연구자들이 업로드한 raw 데이터라고 한다면 Datasets (GDSxxx)는 관리자들에 의해 큐레이션된 데이터로 볼 수 있습니다. 브라우져를 통해 쉽게 검색할 수 있습니다. Bioconductor에서는 GEOquery라는 패키지로 관련 파일들을 다운로드 받을 수 있습니다.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GEOquery")
library(GEOquery)
#browseVignettes("GEOquery")The GDS class
gds <- getGEO(filename=system.file("extdata/GDS507.soft.gz",package="GEOquery"))
class(gds)
methods(class=class(gds))
Table(gds)
Columns(gds)The GSM class - 샘플의 실제 측정값과 실험 조건 등 샘플별 정보 포함. 참고로 MAS 5.0 알고리즘은 서열의 Perfect-Match (PM)과 Mismatch (MM)를 이용해서 유전자의 발현을 정량화 하는 방법으로 (logged) PM-MM의 평균으로 계산함.
gsm <- getGEO(filename=system.file("extdata/GSM11805.txt.gz",package="GEOquery"))
methods(class=class(gsm))
head(Meta(gsm))
Table(gsm)
Columns(gsm)The GPL class - 사용된 칩의 기본 Annotation 정보
gpl <- getGEO(filename=system.file("extdata/GPL97.annot.gz",package="GEOquery"))
gplThe GSE class - 관련된 샘플, annotation 들의 집합 (실험)
14.2 ExpressionSet class
Biobase 패키지는 지놈 데이터를 관리하기 위한 표준화된 데이터 구조 class인 ExpressionSet를 제공합니다. ExpressionSet은 HT assay 데이터와 실험 meta를 포함하고 있습니다.
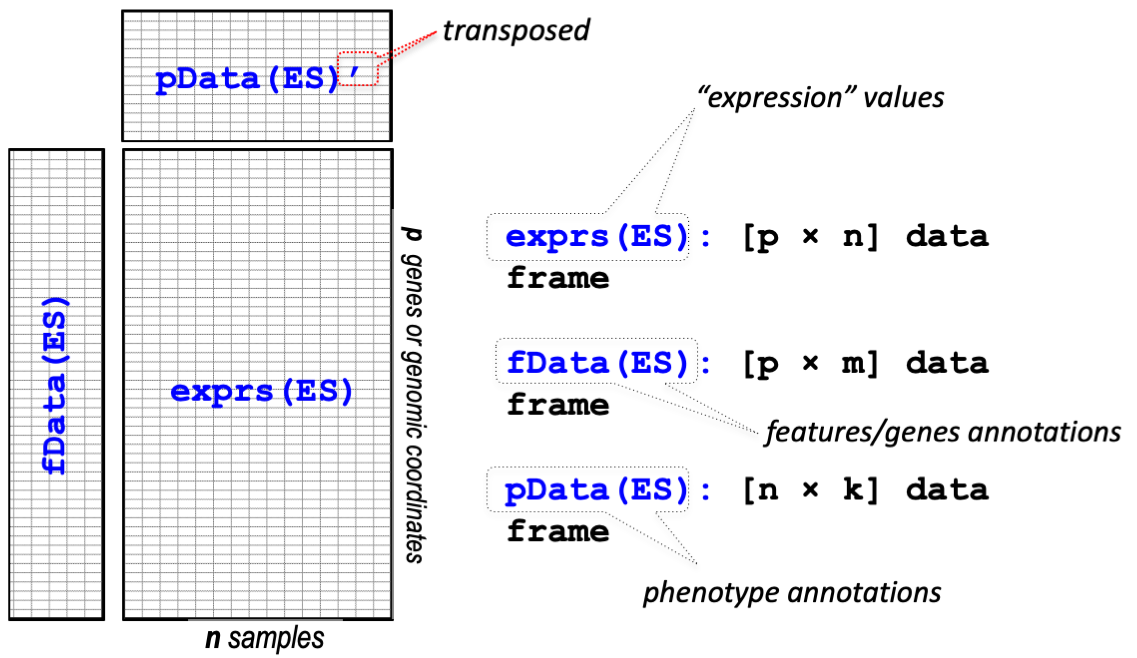 출처
출처GES 데이터 받기 GSE2553
gse2553 <- getGEO('GSE2553',GSEMatrix=TRUE)
gse2553
class(gse2553)
class(gse2553[[1]])
mygse <- gse2553[[1]]
?ExpressionSet
methods(class=class(mygse))
mypdata <- pData(mygse)
myfdata <- fData(mygse)
myexdata <- exprs(mygse)
## rownames , data.frame
as.data.frame(myexdata)
## filtering
table(mypdata$description)
mypdata
class(myexdata)GDS 데이터를 ExpressionSet class로 변환하기
gds <- getGEO(filename=system.file("extdata/GDS507.soft.gz",package="GEOquery"))
class(gds)
eset <- GDS2eSet(gds, do.log2=TRUE)
eset
pData(eset)14.3 ExpressionSet Example
다음 예제는 GEOquery 패키지에 있는 데이터셋을 활용하여 간단한 DEG 분석을 수행하는 코드로서 DEG 분석의 원리 이해와 해석을 위해 학습하는 예제로 볼 수 있습니다. 다음 장에 소개되는 통계(추정과 검정)을 먼저 참고하고 실습해 보아도 좋겠습니다.
GEOquery 패지키에 포함된 GDS507 dataset을 읽어서 ExpressionSet으로 변환합니다. ExpressionSet class를 활용하는 edgeR, DESeq2 등의 패키지를 사용하지는 않지만 변환 후 필요한 데이터를 추출해서 사용하겠습니다.
gds <- getGEO(filename = system.file("extdata/GDS507.soft.gz",package="GEOquery"))
gds
eset <- GDS2eSet(gds, do.log2=TRUE)
eset
myexp <- data.frame(exprs(eset))
myfeature <- fData(eset)
mypheno <- pData(eset)
table(mypheno$disease.state)
class(myexp)
## transformation
mydat1 <- myexp %>%
rownames_to_column() %>%
pivot_longer(cols = -rowname) %>%
pivot_wider(names_from = rowname, values_from = value)
mydat2 <- mypheno %>%
dplyr::select(sample, disease.state) %>%
left_join(mydat1, by = c("sample" = "name"))
mymean <- mydat2 %>%
group_by(disease.state) %>%
summarise(across(where(is.numeric), mean))
mymean2 <- mymean %>%
pivot_longer(-disease.state) %>%
pivot_wider(names_from=disease.state, values_from = value)
ggplot(mymean2, aes(x=normal, y=RCC)) +
geom_point()
이 저작물은 크리에이티브 커먼즈 저작자표시-비영리-변경금지 4.0 국제 라이선스에 따라 이용할 수 있습니다.