11 Biostrings
High-throughput sequencing 데이터를 포함한 DNA나 Amino acid와 같은 생물학적 서열은 Bioconductor의 다양한 패키지들에 의해서 분석될 수 있으며 특히 Biostrings 패키지는 생물학적 서열을 효과적으로 활용하기 위한 핵심 도구로 활용됩니다.
11.1 Working with sequences
Biostrings는 DNA, RNA, amino acids와 같은 생물학적 string을 다루기 위한 다양한 함수를 제공하는 패키지 입니다. 특히 서열에서의 패턴 탐색이나 Smith-Waterman local alignments, Needleman-Wunsch global alignments 등의 서열 비교함수를 제공하여 간단한 서열 분석에 자주 활용되는 패키지 입니다.2 Biostrings 패키지의 설치 방법은 아래와 같습니다.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("Biostrings")Biostrings 패키지는 기본적으로 XString, XStringSet, XStringViews 3가지의 class를 정의하고 있습니다. XString은 DNA나 RNA, AA 등 생물학적 서열 한 가닥을 다루기위한 클래스이며 XStringSet은 여러 가닥을 다루기위한 클래스 입니다.
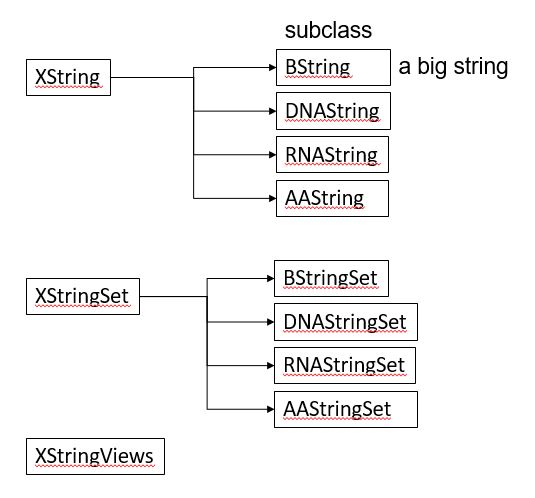
DNAString 함수를 이용해서 객체를 만들어낼 수 있으며 ‘A’, ‘C’, ‘G’, ‘T’ 외에 ‘-’ (insertion), ‘N’ 을 허용합니다.
dna1 <- DNAString("ACGT?")
dna1 <- DNAString("ACGT-N")
dna1[1]
dna1[2:3]
dna2 <- DNAStringSet(c("ACGT", "GTCA", "GCTA"))
dna2[1]
dna2[[1]]
dna2[[1]][1]다음 내장변수 들은 Biostrings 패키지를 로드하면 자동으로 저장되는 변수들로 생물학적 서열을 미리 정의해 놓았습니다. IUPAC (International Union of Pure and Applied Chemistry, 국제 순수·응용 화학 연합)
DNA_BASES
DNA_ALPHABET
IUPAC_CODE_MAP
GENETIC_CODE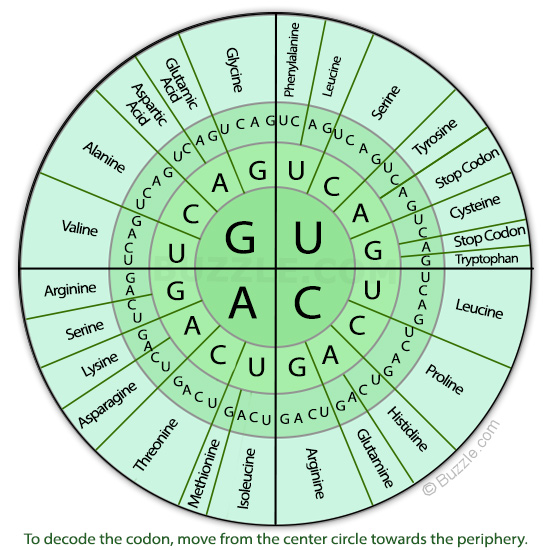
위 변수들을 이용하면 다음처럼 sample() 함수를 이용해서 랜덤하게 DNA 서열을 얻을 수 있습니다. DNA_BASES가 4개 길이를 갖는 벡터인데 이 중 10개를 뽑으려면 replace=T로 해야 합니다.
x0 <- sample(DNA_BASES, 10, replace = T)
x0
s1 <- "ATG"
s2 <- "CCC"
s3 <- paste(s1, s2, sep="")
s3
x1 <- paste(x0, collapse="")
x1관련 함수는 Cheat sheat 참고
11.1.1 XString
XString 클래스는 DNAString과 RNAString, AAString의 subclass로 나눌 수 있습니다. DNAString class에서 length 함수는 핵산의 갯수를 (DNAStringSet 타입의 변수에서 length는 DNA 가닥의 갯수) 계산하며 핵산의 갯수는 nchar함수로 얻어낼 수 있습니다. toString은 DNAString 타입을 단순 문자열로 변환해주는 함수이며 상보서열, 역상보서열 등의 정보도 complement, reverseComplement 등을 사용하여 찾아낼 수 있습니다.
x0 <- paste(sample(DNA_BASES, 10, replace = T), collapse="")
x1 <- DNAString(x0)
class(x0)
class(x1)
length(x1)
toString(x1)
complement(x1)
Biostrings::complement(x1)
reverseComplement(x1)DNAString의 인덱싱은 vector (string)과 같으며 DNAStringSet은 list의 인덱싱과 같습니다.
## indexing
x1[1]
x1[1:3]
subseq(x1, start=3, end=5)
subseq(x1, 3, 5)
## letter frequency
alphabetFrequency(x1, baseOnly=TRUE, as.prob=TRUE)
letterFrequency(x1, c("G", "C"), as.prob=TRUE)Exercises
개시코돈과 스탑코돈을 포함한 30개 길이를 갖는 랜덤 유전자서열을 하나 만드시오
AA_ALPHABET은 IUPAC에서 정의된 아미노산 서열 알파벳이 저장된 내장변수임. “M”과 “*“를 포함하는 10개 길이를 갖는 랜덤 유전자서열을 하나 만드시오
11.1.2 XStringSet
XStringSet역시 DNAStringSet, RNAStringSet, 그리고 AAStringSet으로 나눌 수 있으며 DNAStringSet class는 여러개의 DNAString 을 모아 놓은 집합이라고 보면 됩니다. length 함수는 DNA string의 갯수이며 width 또는 nchar 함수로 각 string의 길이를 구할 수 있으며 이 외 대부분의 DNAString 에서 사용되는 함수가 동일하게 사용될 수 있습니다.
x0 <- c("CTC-NACCAGTAT", "TTGA", "TACCTAGAG")
x1 <- DNAStringSet(x0)
class(x0)
class(x1)
names(x1)
names(x1) <- c("A", "B", "C")
length(x1)
width(x1)
subseq(x1, 2, 4)
x1[[1]]
x1[1]
x3 <- DNAString("ATGAGTAGTTAG")
x4 <- c(x1, DNAStringSet(x3))
x4[-1]
x4
alphabetFrequency(x1, baseOnly=TRUE, as.prob=TRUE)
letterFrequency(x1, c("G", "C"), as.prob=TRUE)
rowSums(letterFrequency(x1, c("G", "C"), as.prob=TRUE))
subseq(x4, 2, 4)RNA나 아미노산 역시 동일한 방식으로 적용 가능하며 c 함수를 이용해서 XStringSet으로 변환 가능합니다.
x1 <- paste(sample(AA_ALPHABET, 10, replace = T), collapse="")
x2 <- paste(sample(AA_ALPHABET, 10, replace=T), collapse="")
x3 <- AAString(x1)
x4 <- AAString(x2)
AAStringSet(c(x1, x2))
AAStringSet(c(x3, x4))Exercises
시작코돈과 종결코돈이 있는 길이 36bp 짜리 DNA (랜덤) 서열을 하나 만드시오
위와 같은 랜덤서열 10개 만들어서 DNAStringSet으로 변환하시오
아래는 가장 직관적으로 생각할 수 있는 for를 이용한 방법입니다. 즉, 10개 저장소를 갖는 x0 변수를 미리 생성해 두고 for 문을 돌면서 서열을 하나씩 만들어 저장하는 방법입니다.
x0 <- rep("", 10)
for(i in 1:length(x0)){
tmp <- paste(sample(DNA_BASES, 30, replace = T), collapse="")
x0[i] <- paste("ATG", tmp, "TAG", sep="")
}
x0위 코드를 함수로 만들어 보겠습니다. random dna를 만들 때 길이만 다를뿐 같은 코드를 반복해서 사용하고 있습니다. 이럴 경우 DNA 길이를 사용자가 정해주도록 input parameter로 하고 해당 파라메터를 받아 DNA를 만들어 주는 함수를 만들어 사용하면 편리합니다.
data(DNA_BASES)
random_dna <- function(len){
tmp <- paste(sample(DNA_BASES, len, replace = T), collapse="")
x0 <- paste("ATG", tmp, "TAG", sep="")
return(x0)
}
random_dna(len=30)
random_dna(len=40)파라메터로 넘겨진 len 값이 sample 함수의 len에 사용된 것을 참고하세요.
이제 길이 30bp짜리 10개의 서열을 반복해서 만들 때 위 함수를 앞서와 같이 for문을 이용하여 10번 반복해서 실행해 주면 같은 결과를 얻습니다. 위와 같이 함수를 만들어 두면 언제든 DNA 서열을 만들 때 재사용 할 수 있습니다.
그런데 R에는 apply 와 같은 행렬연산 함수가 있어서 for문을 사용하지 않고 편리하게 반복문을 실행할 수 있습니다. replicate 함수는 apply와 같은 기능으로 list나 vector 변수에 대해서 사용할 수 있습니다. 즉, 다음과 같이 사용자가 원하는 함수를 반복해서 실행하고 반복 수 만큼의 길이를 갖는 결과를 반환합니다.
x0 <- replicate(10, random_dna(30))
x0
x1 <- DNAStringSet(x0)
x1- 위 생성한 10개 서열의 GC 비율을 계산하고 bar그래프를 그리시오
위 x0 스트링들을 XStringSet으로 바꾸고 GC 비율을 구한 후 bargraph를 그리겠습니다. gc_ratio가 G와 C의 비율값을 저장한 10x2 테이블이므로 x축에 10개의 서열과 각 서열의 GC비율을 나타내고 y축에 비율 값을 그리는 것으로 생각한 후 ggplot의 aes와 파라메터를 적절히 지정해 줍니다.
x1 <- DNAStringSet(x0)
gc_ratio1 <- letterFrequency(x1, c("G", "C"), as.prob=TRUE)
gc_ratio2 <- rowSums(gc_ratio1)
barplot(gc_ratio2, beside=T)
names(gc_ratio2) <- paste("seq", 1:length(gc_ratio2), sep="")
barplot(gc_ratio2, beside=T)
data.frame(gc_ratio2) %>%
rownames_to_column() %>%
ggplot(aes(x=rowname, y=gc_ratio2, fill=rowname)) +
geom_bar(stat="identity") +
scale_y_continuous(limits = c(0, 1)) +
scale_fill_brewer(palette = "green") +
theme_bw()11.1.3 XStringView
Biostrings의 또 다른 class인 XStringView는 XString class의 DNA, RNA, AA서열을 사용자가 원하는대로 볼 수 있는 인터페이스를 제공합니다. 사용법은 다음과 같습니다.
x2 <- x1[[1]]
Views(x2, start=1, width=20)
Views(x2, start=1, end=4)
Views(x2, start=c(1,3), end=4)
Views(x2, start=c(1,3,4), width=20)
Views(x2, start=c(1,3,4), width=20)
i <- Views(x2, start=c(1,3,4), width=20)다음과 같이 한 서열에 대한 여러 부분의 서열 조각도 볼 수 있으며 gaps 함수는 매개변수로 주어진 서열 view의 구간을 제외한 나머지 구간의 서열을 보여주는 함수입니다. successiveviews 함수는 처음 서열부터 매개변수 width에 주어진 갯수 만큼의 서열을 보여주며 rep() 함수를 이용해서 서열의 처음부터 끝까지 보여주는 기능을 합니다.
v <- Views(x2, start=c(1,10), end=c(3,15))
gaps(v)
successiveViews(x2, width=20)
successiveViews(x2, width=rep(20, 2))
successiveViews(x2, width=rep(20, 3))Exercises
- 1000bp 길이의 랜덤 DNA 서열을 만들고 40bp 단위의 길이로 보는 코드를 작성하시오.
앞서 만들어둔 random_dna() 함수를 사용하면 되며 successiveViews 함수를 사용해야 하므로 DNAString으로 변환이 필요하며 서열의 길이에 따라서 rep() 를 이용하여 반복 횟수를 자동 계산합니다.
11.2 Sequence read and write
Biostrings 패키지의 readDNAStringSet이나 writeXStringSet을 사용하면 기본 DNA/RNA/AA 서열의 읽고 쓰기가 가능하며 fasta와 fastq 등의 파일타입으로 적용이 가능합니다.
x1 <- DNAStringSet(x0)
writeXStringSet(x1, "myfastaseq.fasta", format="fasta")
names(x1) <- "myfastaseq"
writeXStringSet(x1, "myfastaseq.fasta", format="fasta")
myseq <- readDNAStringSet("myfastaseq.fasta", format="fasta")
myseqsuccessiveViews로 나눈 여러개의 DNA 조각을 myfastaseqs.fasta에 저장하고 다시 읽을 수 있습니다.
myseqs <- DNAStringSet(sv)
names(myseqs) <- paste("myseqs", 1:length(myseqs), sep="")
writeXStringSet(myseqs, "myfastaseqs.fasta", format="fasta")11.3 Sequence statistics
oligonucleotideFrequency 는 width와 step 이라는 옵션에 따라서 해당 서열의 모든 핵산의 수를 세어주는 합수입니다. 다음에 사용되는 yeastSEQCHR1는 Biostrings 패키지에 포함된 내장 데이터로서 yeast의 첫 번째 염색체 정보를 담고 있습니다.
data(yeastSEQCHR1) #Biostrings
yeast1 <- DNAString(yeastSEQCHR1)
oligonucleotideFrequency(yeast1, 3)
dinucleotideFrequency(yeast1)
trinucleotideFrequency(yeast1)
tri <- trinucleotideFrequency(yeast1, as.array=TRUE)
tri아미노산 정보를 얻기 위해서 ORF를 찾아보겠습니다. yeast의 첫 번째 염색체에 대한 정보는 annotation이 되어 있지만 학습을 위해 툴을 사용하겠습니다. 이미 많은 종류의 ORF 탐색 툴이 나와있지만 본 강의에서는 NCBI에서 제공하는 orffinder를 사용하도록 하겠습니다.
my_ORFs <- readDNAStringSet("yeast1orf.cds")
hist(nchar(my_ORFs), br=100)
codon_usage <- trinucleotideFrequency(my_ORFs, step=3)
global_codon_usage <- trinucleotideFrequency(my_ORFs, step=3, simplify.as="collapsed")
colSums(codon_usage) == global_codon_usage
names(global_codon_usage) <- GENETIC_CODE[names(global_codon_usage)]
codonusage2 <- split(global_codon_usage, names(global_codon_usage))
global_codon_usage2 <- sapply(codonusage2, sum) yeast 첫 번째 염색체에 대한 정보는 bioconductor annotationData OrgDb 또는 bioconductor annotationData TxDb 에서 찾아볼 수 있습니다.
#BiocManager::install("org.Sc.sgd.db")
library(org.Sc.sgd.db)
class(org.Sc.sgd.db)
?org.Sc.sgd.db
ls("package:org.Sc.sgd.db")
columns(org.Sc.sgd.db)
mykeys <- keys(org.Sc.sgd.db, keytype = "ENTREZID")[1:10]
AnnotationDbi::select(org.Sc.sgd.db,
keys=mykeys,
columns = c("ORF","DESCRIPTION"),
keytype="ENTREZID")TxDb
BiocManager::install("TxDb.Scerevisiae.UCSC.sacCer3.sgdGene")
library(TxDb.Scerevisiae.UCSC.sacCer3.sgdGene)
class(TxDb.Scerevisiae.UCSC.sacCer3.sgdGene)
columns(TxDb.Scerevisiae.UCSC.sacCer3.sgdGene)
methods(class=class(TxDb.Scerevisiae.UCSC.sacCer3.sgdGene))
ygenes <- genes(TxDb.Scerevisiae.UCSC.sacCer3.sgdGene)
library(tidyverse)
mydat <- global_codon_usage2 %>%
data.frame %>%
rownames_to_column %>%
rename(codon = "rowname", freq = ".")
ggplot(mydat, aes(x=codon, y=freq)) +
geom_bar(stat="identity")
mydat
AMINO_ACID_CODE[mydat$codon]Exercises
AMINO_ACID_CODE를 이용해서 위 그래프의 1약자를 3약자로 변환, 라벨을 세로로 90도 회전, y축 라벨 “Frequency”, x축 라벨 “Amino acid code”, theme 옵션 “theme_bw” 등을 적용하여 다시 그림을 그리시오 (revisit ggplot2)
11.4 Pattern matching
Biostrings 패키지에는 하나의 subject 서열에 특정 pattern이 존재하는지 탐색하는 matchPattern함수를 제공합니다. 만약 여러개의 subject 서열에서 하나의 pattern을 찾을 경우에는 vmatchPattern함수를 사용하고 하나의 subject 서열에 여러개의 pattern을 찾는 경우에는 matchPDict 함수를 사용합니다.
length(coi)
hits <- matchPattern("ATG", yeast1, min.mismatch=0, max.mismatch=0)
hits
class(hits)
methods(class="XStringViews")
ranges(hits)
hits <- vmatchPattern("ATG", my_ORFs, min.mismatch=0, max.mismatch=0)
stack(hits)
이 저작물은 크리에이티브 커먼즈 저작자표시-비영리-변경금지 4.0 국제 라이선스에 따라 이용할 수 있습니다.