16 High-throughput data II
16.1 SummarizedExperiment Class
ExpressionSet은 일반적으로 행이 feature (유전자) 인 마이크로어레이 기반 실험 및 유전자 발현 데이터에 사용되었습니다. 그러나 유전체 분석을 위해서는 유전자 정보 외에도 유전체상의 위치 정보 등이 필요하며 이는 앞서 배운 GenomicRanges 형태의 데이터가 필요합니다. 따라서 최근에는 새로운 버전인 SummarizedExperiment class가 SummarizedExperiment 개발되어 사용되고 있습니다.
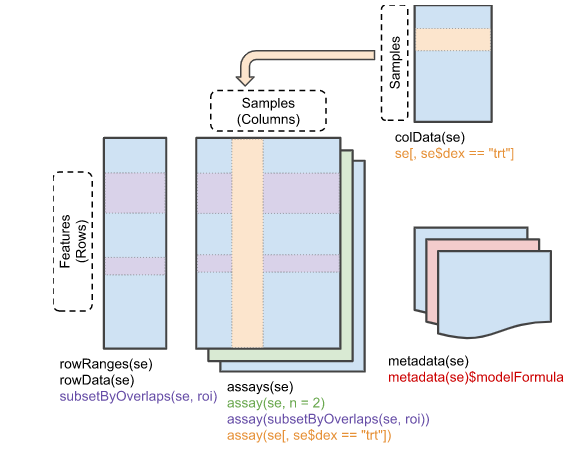
library(SummarizedExperiment)
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("airway")
library(airway)
data(airway, package="airway")
se <- airway
se
?RangedSummarizedExperiment
# Row (features)
rowRanges(se)
# Column (sample)
colData(se)
# Experiment-wide metadata
metadata(se)시퀀싱 파일은 GEO 데이터베이스 GSE52778에 있으며 패키지에도 기본으로 포함되어 있습니다.
dir <- system.file("extdata", package="airway", mustWork=TRUE)
list.files(dir)
readLines(file.path(dir, "Homo_sapiens.GRCh37.75_subset.gtf")).bam 파일은 RNA-Seq read를 포함하고 있으며 .csv 파일은 실험 디자인, .gtf 파일은
- gff 파일 포맷과 gtf 파일 포멧 차이
csvfile <- file.path(dir, "sample_table.csv")
sampleTable <- read.csv(csvfile, row.names = 1)
bamfilenames <- file.path(dir, paste0(sampleTable$Run, "_subset.bam"))Rsamtools는 bam이나 sam 파일을 읽을 수 있는 패키지로 yieldSize 파라메터로 메모리 과사용을 막을 수 있습니다.
library(Rsamtools)
bamfiles <- BamFileList(bamfilenames, yieldSize = 2000)
bamfiles
class(bamfiles)
seqinfo(bamfiles[1])bam 파일 내의 read들은 염색체 서열의 각 위치에 mapping 되어 있으며 유전자의 발현을 보기 위해서는 각 유전자에 mapping 된 read들의 수를 세어야 합니다.
SummarizedExperiment 생성
nrows <- 200
ncols <- 6
counts <- matrix(runif(nrows * ncols, 1, 1e4), nrows)
rowRanges <- GRanges(rep(c("chr1", "chr2"), c(50, 150)),
IRanges(floor(runif(200, 1e5, 1e6)), width=100),
strand=sample(c("+", "-"), 200, TRUE),
feature_id=sprintf("ID%03d", 1:200))
colData <- DataFrame(Treatment=rep(c("ChIP", "Input"), 3),
row.names=LETTERS[1:6])
se <- SummarizedExperiment(assays=list(counts=counts),
rowRanges=rowRanges, colData=colData)
# Row (regions-of-interest) data
rowRanges(se)
# Column (sample) data
colData(se)
# Experiment-wide metadata
metadata(se)16.2 High-throughput genomic data analysis
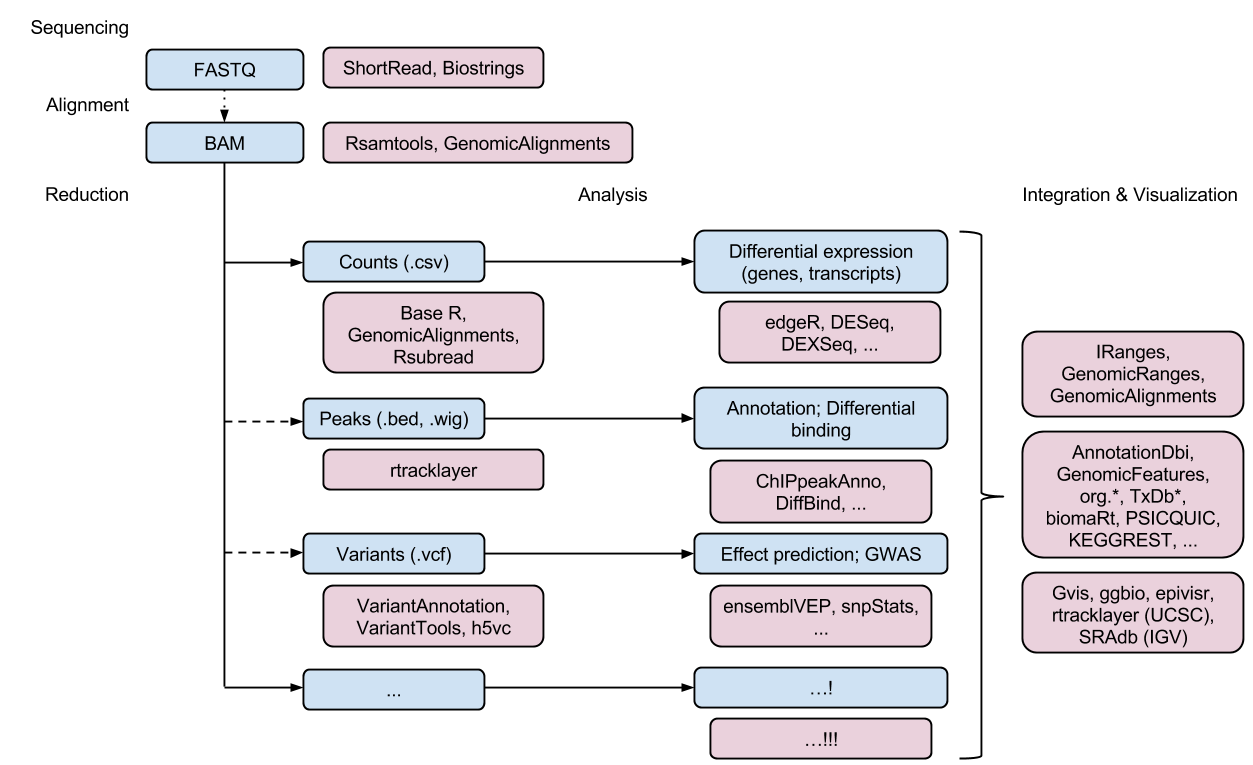
- Bioconductor에는 RNA-seq를 포함한 대규모 high-throughput 서열 데이터의 분석을 지원하는 많은 패키지가 있습니다.
본 학습에서는 아래 소개된 분석 예제를 통해 RNA-Seq 데이터 분석의 기술을 익히도록 합니다. RNA-Seq CSAMA 2022. 사용되는 데이터는 airway2 에 저장된 8개의 RNA-seq 샘플에 (1차 인간 기도 평활근 세포주 4개 처리군, 4개 대조군) 대한 정량 데이터가 되겠으며 PMID: 24926665. GEO: GSE52778 및 다음 논문을 참고하시면 되겠습니다. (Himes BE, Jiang X, Wagner P, Hu R, Wang Q, Klanderman B, Whitaker RM, Duan Q, Lasky-Su J, Nikolos C, Jester W, Johnson M, Panettieri R Jr, Tantisira KG, Weiss ST, Lu Q. ‘RNA-Seq Transcriptome Profiling Identifies CRISPLD2 as a Glucocorticoid Responsive Gene that Modulates Cytokine Function in Airway Smooth Muscle Cells.’ PLoS One. 2014 Jun 13;9(6):e99625)
library(airway)
dir <- system.file("extdata", package = "airway")
list.files(dir)
list.files(file.path(dir, "quants"))16.3 rtracklayer
https://www.bioconductor.org/packages/release/bioc/vignettes/rtracklayer/inst/doc/rtracklayer.pdf
rtracklayer: UCSC web-based genome browser
16.4 ggbio
Make gene model from OrganismDb object
library(ggbio)
library(Homo.sapiens)
class(Homo.sapiens)
data(genesymbol, package = "biovizBase")
wh <- genesymbol[c("BRCA1", "NBR1")]
wh <- range(wh, ignore.strand = TRUE)
p.txdb <- autoplot(Homo.sapiens, which = wh)
p.txdb
# orgDB
library(org.EcK12.eg.db)
class(org.EcK12.eg.db)
methods(class="OrgDb")
columns(org.EcK12.eg.db)
keys(org.EcK12.eg.db)
tmp <- AnnotationDbi::select(org.EcK12.eg.db, keys(org.EcK12.eg.db), "ACCNUM")
org.EcK12.eg.db$ACCNUM- BSgenome DB 보다 최신 정보를 얻기 위해서는 NCBI 직접 다운로드
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("BSgenome.Ecoli.NCBI.20080805")
library(BSgenome.Ecoli.NCBI.20080805)
?BSgenome.Ecoli.NCBI.20080805
genome <- BSgenome.Ecoli.NCBI.20080805
seqlengths(genome)
available.genomes()
genome$NC_008253
library(rentrez)
library(genbankr)
tmps <- entrez_fetch("nuccore", id="NC_000913.3", rettype="gbwithparts")
write(tmps, "ecoli-mg1655.gb")
ecoligb <- readGenBank("ecoli-mg1655.gb")
ecoli_cds <- cds(ecoligb)
ecoli_cds
p.txdb <- autoplot(ecoli_cds)
p.txdb
#library(igvR)
ecoli_cds
ggbio() +
circle(ecoli_cds, geom = "ideo", fill = "gray70") +
circle(ecoli_cds, geom = "scale", size = 5) +
circle(ecoli_cds, geom = "text", aes(label = locus_tag), vjust = 0, size = 3) +
theme(
axis.text.x = element_text(angle=90)
)
gr1 <- granges(ecoli_cds)
gr2 <- granges(ecoli_cds)
mcols(gr2)$test <- rnorm(length(ecoli_cds))
ggplot() +
layout_circle(ecoli_cds, geom = "ideo", fill = "gray70", radius = 9, trackWidth = 1) +
layout_circle(ecoli_cds, geom = "scale", size = 3, trackWidth = 1, scale.n=20) +
layout_circle(gr1, geom = "rect", color = "steelblue", radius = 5) +
layout_circle(gr2, geom = "bar", aes(y=test), size = 3, trackWidth = 1, scale.n=20, radius = 4)
library(igvR)
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("igvR")
igv <- igvR()
setGenome(igv, "hg38")16.5 Practice for review
NC_000913.3은 미생물 연구에서 잘 알려진 Escherichia coli str. K-12 substr. MG1655 균주의 Reference sequence 입니다. 해당 지놈 서열의
fasta와gb형태의 포맷으로rentrez패키지를 사용해서 다운로드 하고ecoli-k12.fasta,ecoli-k12.gb파일로 각각 저장하시오. 참고로 genbank 포맷의 full version은rettype=옵션을gbwithparts로 해야함.genbankr패키지를 사용해서ecoli-k12.gb파일을 읽어들이고 Coding sequence 를 추출해서ecolicds라는 변수에 저장하시오plyranges패키지의 filter 함수를 이용해서 ompR 이라는 유전자가 있는지 찾으시오ecolicds에서 다음 12개의 TF에 대한 정보를 추출하시오
"araC" "sgrR" "leuO" "cra" "mraZ" "pdhR" "cdaR" "rclR" "betI" "pdeL" "cynR" "lacI"-
NC_010572.1는 celR이라는 전사인자 단백질로서 cellulase 분해 유전자들의 발현을 제어함. 다음 서열을
celR.fasta파일로 저장하고Biostrings패키지를 사용해서 읽어들이시오.
>NC_010572.1 celR Streptomyces griseus subsp.
ATGGCGGCATCGCGAGTACGGAACGGCGGGCGGCCCACGCTCGAAGAGGTCGCGGCACGGGCCGGGGTCG
GCCGGGGCACCGCCTCACGGGTCATCAACGGCTCGCCCCGGGTCAGCGACGCCACCCGGCAGGCCGTCGA
GGCGGCCGTCGCCGAACTGGGGTACGTCCCCAACCGCGCCGCCCGCGCCCTGGCGGGCAACCGCACCGAC
GCCATCGCGCTGGTGGTCCCCGAGCCGGAGACCCGCTTCTTCGCCGAGCCCTACTTCTCCGCCATAGTGC
GCGGTGTCGGGGCGGCCCTGGCCGACACCGAGATGCAGCTGCTCCTCACCCTCGTCGGCAACGACCGCGA
GCGCCGCAGGCTCGCCCAGTACCTCACCGCCCACCGCGTCGACGGGGTCCTCCTGGTCGCCGTGCACGCC
GATGACCCGCTGCCGGAGCTCCTGGAGCAGCTGGGCATGCCCTGCGTGATCAGCGGCGCCCGGCACGCGG
CCGAGACGCTGCCCTCGGTCGACTCCGACAACTTCGAGGGCGCGCGGGCCGCCGTGGAGCACCTGGTCTC
CCGGGGCCGCCGCCAGGTGGCCACCATCACCGGCCGCCTGGAGGTCTACGGCGCCCAGCGCCGCCTGGAC
GGCTACCGCGCCGCGGTCTCCGCCGCCGGCCTGGCCCCCGACGAGCGCCTGATCGCCCCGGCCGACTTCA
CCGAGGAGGGCGGCGCCCGGGCCATGCGCGACCTCCTGGCCCGCCGCCCCGGCCTCGACGCCGTCTTCGT
GGCCTCCGACGTGATGGCCGCGGGCGCCCGCCAGGTCCTGCGCGAGGCGGACCGCCGCATCCCCGAGGAC
GTGGCCCTGATCGGCTTCGACGACTCGGTGGTCGCCCGCCACATGCACCCGGCCCTCACCAGCGTCCGCC
AGCCCATCGAGGAGATGGGCCGCCGGATGGCCCAGCTCCTCCTGGACGAGATCGCGGGCCGGGCCCCGGG
CGACGAGCGCCCCTCGGTGGTCCTGCCCACGGAGCTGGTGGTCCGCGACTCGTCGTGA- 앞서 ecoli의 전체 지놈 서열을 추출하고 ecoli 지놈에 celr 서열과 유사 서열이 있는지
Biostrings의pairwiseAlignment함수를 이용해서 탐색하시오
16.6 BLAST on local machine
일반적으로 유사 서열을 탐색할 경우 ncbi의 blast을 사용합니다. rBLAST는 BLAST local을 설치한 컴퓨터에서 R을 활용해서 blast를 수행할 수 있게 만든 패키지입니다. local blast 설치는 BLAST Command Line Applications User Manual을 참고하거나 docker 이미지, docker blast manual을 사용해도 되겠습니다.
docker pull ncbi/blast
docker images
library(Biostrings)
dbfile <- "ecolicdsseq.fasta"
ecolicdssec <- getSeq(ecoliseq, ecolicds)
writeXStringSet(ecolicdssec, dbfile)
targetfile <- "celrseq.fasta"
writeXStringSet(celr, targetfile)파일탐색기에서 working directory로 이동한 후 cmd 실행. 다음 명령어로 blastn 실행 가능.
docker run --rm -v %cd%:/myhome ncbi/blast blastn 데이터베이스 만들기, dbtype은 핵산의 경우에는 ‘nucl’, 단백질의 경우에는 ‘prot’. %cd%는 윈도우 cmd 환경에서 현재 디렉토리를 나타내는 문자
docker run --rm -v %cd%:/myhome -w /myhome ncbi/blast makeblastdb -in ecolicdsseq.fasta -dbtype nucl -out ecoliblastn 수행
docker run --rm -v %cd%:/myhome -w /myhome ncbi/blast blastn -query celrseq.fasta -db ecoli -out blast_output.txt다음 서열을 ribosomalprot.fasta 파일로 저장
>X02130.1 E. coli genes rpsI and rplM for ribosomal proteins S9 and L13
AACACTCGTCCGAGAATAACGAGTGGATCTTTGACCCCGACTTCTCTATAATCCTGCGACCCCACGTTAC
AAGAAAGTTTTTTTCCCAAAACTTTTTGTGTGCTGGCATAGGCTATTCGAAGGGGTAGGTTTGCCGGACT
TTGTCGTGTGAACCTCAACAATTGAAGACGTTTGGGTGTTCACCAACGTGTAACTATTTATTGGGTAAGC
TTTTAATGAAAACTTTTACAGCTAAACCAGAAACCGTAAAACGCGACTGGTATGTTGTTGACGCGACCGG
TAAAACTCTGGGCCGTCTGGCTACTGAACTGGCTCGTCGCCTGCGCGGTAAGCACAAAGCGGAATACACT
CCGCACGTAGATACCGGTGATTACATCATCGTTCTGAACGCTGACAAAGTTGCTGTAACCGGCAACAAGC
GTACTGACAAAGTGTACTATCACCACACCGGCCACATCGGTGGTATCAAACAAGCGACCTTTGAAGAGAT
GATTGCTCGCCGTCCTGAGCGTGTGATTGAAATCGCGGTTAAAGGCATGTTGCCAAAAGGCCCGCTGGGT
CGTGCTATGTTCCGTAAACTGAAAGTTTACGCGGGTAACGAGCACAACCACGCGGCACAGCAACCGCAAG
TTCTTGACATCTAATCGGATTATAGGCAATGGCTGAAAATCAATACTACGGCACTGGTCGCCGCAAAAGT
TCCGCAGCTCGCGTTTTCATCAAACCGGGCAACGGTAAAATCGTAATCAACCAACGTTCTCTGGAACAGT
ACTTCGGTCGTGAAACTGCCCGCATGGTAGTTCGTCAGCCGCTGGAACTGGTCGACATGGTTGAGAAACT
GGACCTGTACATCACCGTTAAAGGTGGTGGTATCTCTGGTCAGGCTGGTGCGATCCGTCACGGTATCACC
CGCGCTCTGATGGAATACGACGAGTCCCTGCGTTCTGAACTGCGTAAAGCTGGCTTCGTTACTCGTGACG
CTCGTCAGGTTGAACGTAAGAAAGTCGGTCTGCGTAAAGCACGTCGTCGTCCGCAGTTCTCCAAACGTTA
ATTGGCTTCTGCTCCGGCAGAAAACAATTTTCGAAAAAACCCGCTTCGGCGGGTTTTTTTATAGGGAAGG
TGCGAACAAGTCCCTGATATGAGATCATGTTTGTCATCTGGAGCCATAGAACAGGGTTCATCAT
>X04022.1 E. coli genes rpsF, rpsR and rplI for ribosomal proteins S6, S18, L9
CAAGCTTTGCACATCGTCCATATTTCTGGCCTGGTGGTTATTAATTTCAATGGCTGCCCATGTATTTGCA
CTTAGCAAAAGCACAGCCAGAAGGGCTAAAACACGACTGAACATAGATACCTCCTCGACGGCTGACTTTG
TGTGCTCTCCTTCCTCGTGATGATCTTCTCGATTTAATTTTAATCAATGATAAAGAAGTTGATGGTGACC
ATTTCTGATGCAGTTGTTCAAAAAAACCACCATGATGAAGTGTGATGAACTTCAAATCAGCGTGTTAGAG
GTTAATTGCGAAAGGGGAGATTTATTTCGGCTCTGCCCTTGAGTTTAGCGAGGCATACAAGTACTATAAC
GGCGTCATTTTTCAGCCGACCTTTAACACGTTCCTTGCCTCCCCGGGATTCGGCTGACCCAGACAGGAGG
CGTGAATAATCCGTAAGGAGCAATTCGATGCGTCATTACGAAATCGTTTTTATGGTCCATCCTGATCAGA
GCGAACAGGTTCCGGGCATGATCGAGCGCTACACTGCTGCCATCACTGGTGCAGAAGGCAAGATCCACCG
TCTGGAAGACTGGGGCCGCCGTCAGCTGGCTTACCCGATCAACAAACTGCACAAAGCACACTACGTTTTG
ATGAATGTTGAAGCTCCGCAGGAAGTGATCGATGAGCTGGAAACTACCTTCCGCTTCAACGATGCCGTTA
TCCGCAGCATGGTTATGCGTACCAAGCACGCTGTTACCGAAGCATCTCCGATGGTTAAAGCGAAAGACGA
GCGCCGTGAGCGTCGCGATGATTTCGCAAACGAAACCGCTGATGATGCTGAAGCTGGGGATTCTGAAGAG
TAATTTCTGATGACCAACCGTCTGGTGTTGTCCGGCACCGTGTGCAGGGCTCCCCTTCGAAAGGTCAGTC
CATCAGGAATTCCTCACTGCCAGTTCGTGCTTGAGCATCGTTCTGTGCAGGAGGAAGCCGGCTTTCACCG
GCAGGCGTGGTGTCAAATGCCCGTTATTGTTAGCGGACACGAAAACCAGGCCATTACTCACAGTATAACG
GTCGGCAGTCGCATAACCGTTCAGGGGTTCATTTCATGCCACAAGGCAAAGAACGGACTGAGCAAAATGG
TTTTGCATGCCGAGCAGATTGAATTGATAGATTCTGGAGACTAGCCATATGGCACGTTATTTCCGTCGTC
GCAAGTTCTGCCGTTTCACCGCGGAAGGCGTTCAAGAGATCGACTATAAAGATATCGCTACGCTGAAAAA
CTACATCACCGAAAGCGGTAAGATTGTCCCAAGCCGTATCACCGGTACCCGTGCAAAATACCAGCGTCAG
CTGGCTCGCGCTATCAAACGCGCTCGCTACCTGTCCCTGCTGCCGTACACTGATCGCCATCAGTAATCGG
TCACAGGTCCATTAATACGACTTTGAGAGGATAAGGTAATGCAAGTTATTCTGCTTGATAAAGTAGCAAA
CCTGGGTAGCCTGGGTGATCAGGTAAACGTTAAAGCGGGCTATGCTCGTAACTTCCTGGTACCGCAGGGT
AAAGCTGTTCCAGCTACCAAGAAAAACATTGAATTCTTCGAAGCTCGTCGCGCTGAACTGGAAGCTAAAC
TGGCTGAAGTTCTGGCAGCTGCTAATGCTCGCGCTGAGAAAATCAATGCACTGGAAACTGTTACCATCGC
GTCTAAAGCTGGCGACGAAGGTAAACTGTTCGGTTCCATCGGTACTCGCGACATCGCTGACGCTGTAACT
GCAGCTGGCGTTGAAGTGGCTAAGAGCGAAGTTCGTCTGCCGAACGGCGTTCTGCGTACCACTGGCGAAC
ACGAAGTGAGCTTCCAGGTTCACAGCGAAGTATTCGCGAAAGTGATCGTAAACGTAGTAGCTGAATAATT
CGTTATTCAACGAGACGTAAAAAGCGCCCGACCATTGGTCGGCGTTTTGCTTTCTATTTTTCGTCAGGTA
TTAGTTTCGCAAGTAGATC
>J01677.1 E.coli rpmB and rpmG genes coding for ribosomal proteins L28 and L33
GGATTTAACCCGCTATGCGCGATCCTTCGGGATCTTTGTCTGTTCGGGACTTGAGCACATCGCTGAGTCA
GCGTATACTACGCCACCTTTGAGAATCTCGGGTTTGGCATTTGGGCCTGGCAATCGAGAGTTCACAGAAC
TGCGATGACCGGGCTGTAAAGACCTGACGAGGCGCCAATACCCCATACGAAGCTCGAGCTAATTTGATTT
TTGGAGAATAGACATGTCCCGAGTCTGCCAAGTTACTGGCAAGCGTCCGGTGACCGGTAACAACCGTTCC
CACGCACTGAACGCGACTAAACGCCGTTTCCTGCCGAACCTGCACTCTCACCGTTTCTGGGTTGAGAGCG
AGAAGCGTTTTGTCACCCTGCGCGTATCTGCTAAAGGCATGCGTGTAATCGATAAAAAAGGCATCGATAC
AGTTCTGGCTGAACTGCGTGCCCGTGGCGAAAAGTACTAAGTACTTAGAGGAAATAAATCATGGCTAAAG
GTATTCGTGAGAAAATCAAGCTGGTTTCTTCTGCTGGTACTGGTCACTTCTATACCACTACGAAGAACAA
ACGTACTAAGCCGGAAAAACTGGAACTGAAAAAATTCGATCCAGTTGTTCGCCAGCACGTGATCTACAAA
GAAGCGAAAATCAAATAATTCTCGCTTTGATGTAACAAAAAACCCCGCCCCGGCGGGGTTTTTTGTTATC
TGCTTGCCCCCATATTGACTGCATCTGTTCATTCCTGGAGATGCTATGCCTGAATTACCCGAAGdocker run --rm -v %cd%:/myhome -w /myhome ncbi/blast blastn -query ribosomalprot.fasta -db ecoli -out blast_output.txt출력물 분석위한 옵션 설정
docker run --rm -v %cd%:/myhome -w /myhome ncbi/blast blastn -query ribosomalprot.fasta -db ecoli -outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue stitle" -out blast_output.txt출력물을 읽어들여 아래와 같이 각 hit (서열) 별로 서열 및 관련 단백질 title 정리
library(dplyr)
blastout <- read.delim("blast_output.txt", header = F)
tmplist <- blastout %>%
mutate(query=factor(V1)) %>%
group_by(query) %>%
group_split()
seqid <- lapply(tmplist, function(x){paste(x[1,1])}) %>% unlist
hit_seqid <- lapply(tmplist, function(x){paste(x[1,2])}) %>% unlist
hit_evalue <- lapply(tmplist, function(x){paste(x[1,11])}) %>% unlist
hit_prot_title <- lapply(tmplist, function(x){paste(x[1,12])}) %>% unlist
hitdat <- data.frame(seqid, hit_seqid, hit_prot_title, hit_evalue)
write.table(hitdat, file = "target_blastout_table.tab", sep="\t", quote = F, row.names = F)16.7 Bioconductor Workflow (link)
Bioconductor에서는 다양한 생물학적 데이터를 분석하기 위한 툴이 개발되고 있으며 이러한 툴들은 RNA-seq과 같은 특정 목적을 위해 반복적으로 또는 순차적으로 수행되어야 하고 이러한 일련의 툴 사용 방법을 workflow로 만들어 제공하고 있습니다. 일부 워크플로에 대해서 간단히 리뷰하며 마치도록 하겠습니다.
급성 림프구성 백혈병 데이터 (Annotation)
library(ALL)
data(ALL)
ALL
featureData(ALL)
##
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("hgu95av2.db")
library(hgu95av2.db)
browseVignettes("hgu95av2.db")
help(package="hgu95av2.db")
featureNames(ALL)[1:10]
ids <- featureNames(ALL)[1:10]
as.list(hgu95av2ENTREZID[ids])